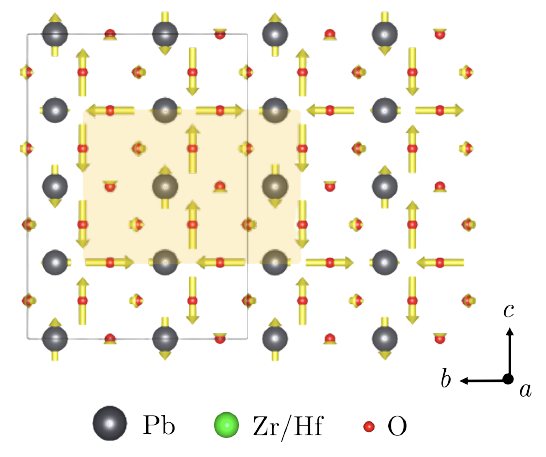
Dipolar vortices in antiferroelectric perovskite materials PbZrO3 and PbHfO3.
Broadly speaking, if aspects of quantum theory can be advantageously used in/applied to a domain, I pursue it. This can be considered the unifying theme of my research interests, bringing together early fault tolerant algorthims for quantum optimization, chemistry and machine learning and large-scale first principles simulations of solids. The latter of these of these topics was covered in my Ph.D. research. I used massively parallel density functional functional theory codes on large supercomputers to simulate ferroelectric and antiferroelectric systems at unprecedented scale. These simulations provided accurate charecterizations of topological polar textures and revealed new phase transition behaviour in antiferroelectrics. I am still involved in this area, but now spend most of my time ideating and implementing new algorithms for early fault tolerant quantum computers with key applications in the simulation of quantum materials. Another exciting horizon I am working towards is the much anticipated joining of classical high performance computing with quantum computation.
Use the below links to jump to research highlights:
Now that noisy intermediate scale quantum (NISQ) computers have been built, I am working towards making existing and future fault tolerant machines more than mere physics experiments. While quantum supremacy has now been demonstrated using NISQ hardware for various sampling experiments, the practical use of this supremacy is limited. Therefore, we must either learn to use the limited number of noisy qubits we have at hand to do something really useful and plan for the early fault tolerant era where algorithms come with strict performance guarantees.
One promizing area is quantum machine learning (QML). I have developed various QML algorithms suited for NISQ and early fault tolerant hardware for time-series analysis, outlier detection, and others utilizing novel hybrid-quantum classical neural network architectures and multiple kernel learning. Although practical quantum advantage is the end goal, but there are many obstacles along the way. There are, for example, many unsolved problems in fundamental QML research for which I am interested in finding solutions to or at least alleviating including barren plateaus (induced by hardware noise or lack of inductive bias), efficient training of quantum circuits and understanding the generalization bounds of different QML models.
A schematic demonstrating the quantum approximate optimization algorithm applied to portfolio optimization.Another area which intrigues me is the potential for quantum algorithms to approximately solve NP-hard combinatorial optimization problems. Such problems appear everywhere from finance to route optimization so are researched heavily. There are very well-known algorithms for this task (notably the quantum approximate optimization algorithm and quantum annealing) but they face problems at scale. I am therefore interested in (i) enhancing existing approaches using classical deep learning techniques and others (ii) working towards new optimization algorithms in general. My recent research in this area revolves around benchmarking quantum optimization on NISQ hardware for finance applications like portfolio optimization and diversification and exploring new ways for assesing the quality of solutions yielded from these methods.
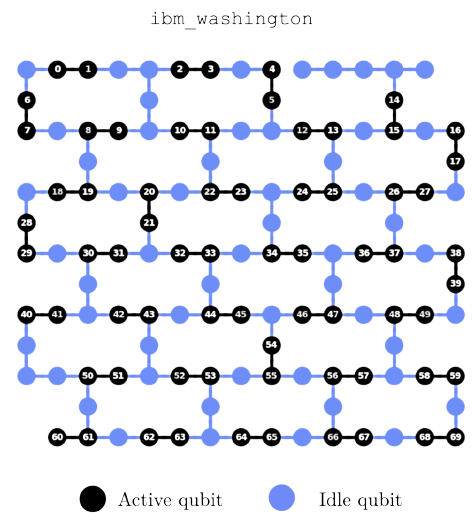
Many parallelized quantum circuits spread throughout the 127 qubit superconducting transmon quantum computer: ibm_washington.A final important topic I will mention is levaraging algorithm-agnostic tricks for either speeding up the execution time of or increasing the quality of results from quantum algorithms. For the former, I investigate new ways of computing parallel quantum circuits on (relatively) large quantum computers using quantum multi-programming. This can be considered a quantum analgoue for well established classical distributed computing paradigms like the Message Passing Interface. For the latter, I study how error mitigation techniques like dynamical decoupling and pauli twirling can help to increase application specific performance in QML and optimization. Furthermore, all of these techniques can be applied in tandem using new hybrid computing systems (like Qiskit Runtime etc.) helping to bring about the intersection of quantum and classical high performance computing.
Polar morphologies from first principles
Polar vortex-antivortex pairs in PbTiO3 films on SrTiO3 substrates. These vector fields were calculated using aotmic sturctures from first principles DFT.Broadly, a polar morphology is a distinct pattern of electrical dipole moments arranged in a solid or nanostructure. These (admittedly visually mesmerizing) objects can give rise to novel materials properties including (but not limited to) negative capacitance and giant electromechanical responses. Some of these properties are the direct result of of their unique topologies. Indeed, topological phases like polar skyrmions, polar waves and chiral bubble domains have all been observed existing within the now widely studied PbTiO3/SrTiO3 heterostructure.
Where do I fit in to all of this? Well, from an atomistic perspective, simulations able to capture these polar morphologies need thousands if not millions of atoms. This puts standard implementations of first principles density functional theory (DFT) well out of reach and leaves most practitioners dealing with second principles methods and phase-field modelling. However, using large-scale first principles DFT methods, we can simulate these systems provided we have huge computational resources (thousands to millions of physical cores) to lean on. This is what I am involved with; massively parallel simulations of polar textures using large supercomputers.
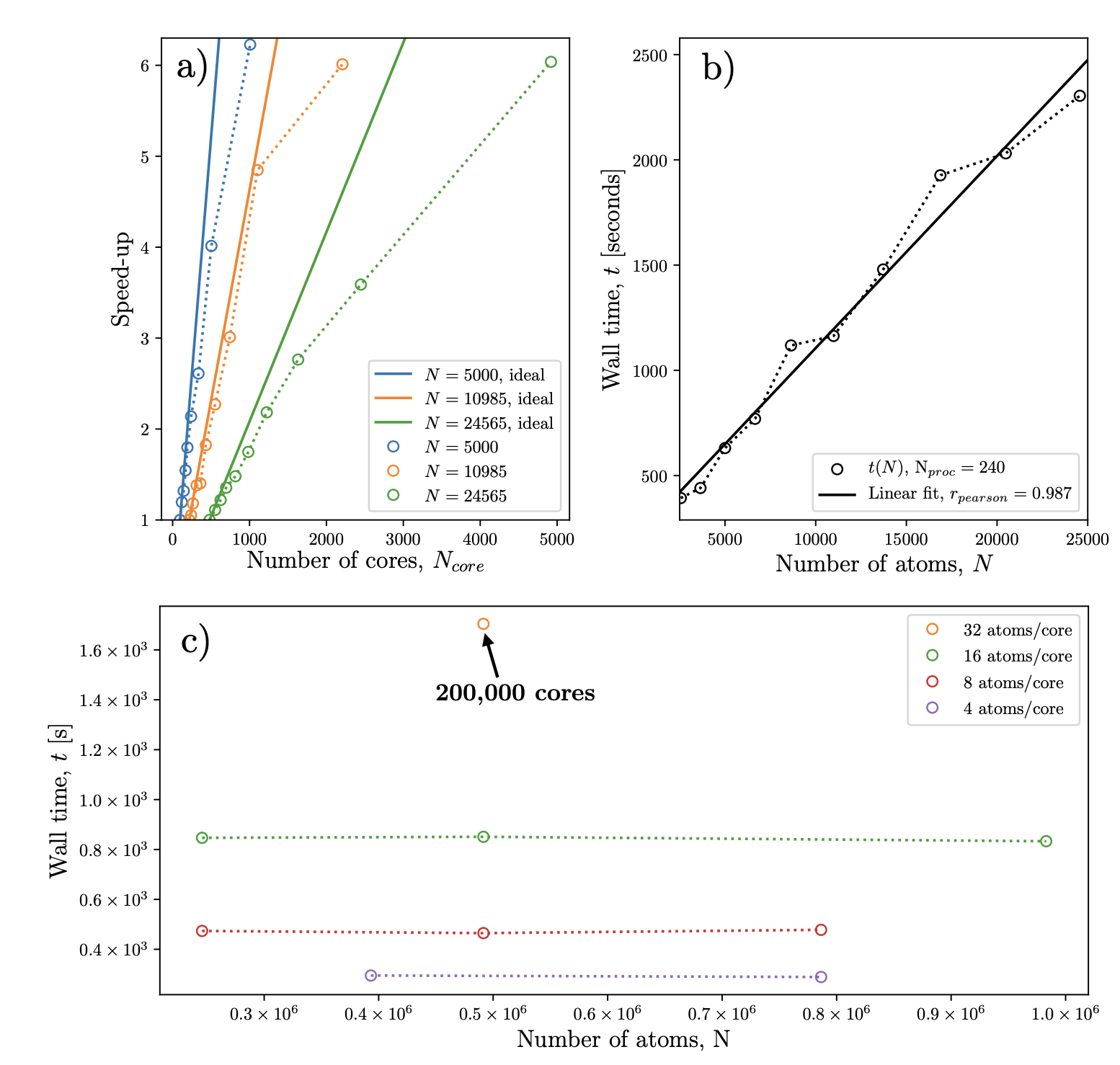
Scaling behaviour of the CONQUEST code for different numbers of atoms and cores using PbTiO3 as a model system. (a) strong scaling behaviour vs ideal. (b) Linear scaling with system size. (c) Weak scaling behaviour with up to 200,000 physical cores.Using these large-scale techniques, I have unveiled the mechanism causing the formation polar-wave textures in thin ferroelectric films and determined critical thicknesses for the formation of polar flux-closure domains. I have also contributed towards answering some long standing questions regarding the orientation of ferroelectric domains in thin films in the presence of surface trenches. Using large-scale DFT, we discovered that the alignment is due to a favorable arrangement of electrical dipole moments which minimize trench-induced depolarizing fields. This mechanism has broad applicability and can be used to engineer other polar textures in various ferroelectric nanostructures. In the future, I am looking towards conducting the first quantum-mechanical simulations of polar skyrmions using large scale DFT and/or other first principles-derived methods. This will require exascale supercomputing.
Phase transitions in antiferroelectrics
Perhaps the most remarkable thing about antiferroelectric (AFE) materials is how little we know about them. Despite antiferroelectricity being discovered over 70 years ago in the perovskite PbZrO3, we neither fully understand the mechanism for the paraelectric to antiferroelectric phase transition nor have we even come to a consensus on a formal definition for antiferroelectricity. There are therefore many areas to expore in the antiferroelectrics and I shall remark on a few related to my own research interests.
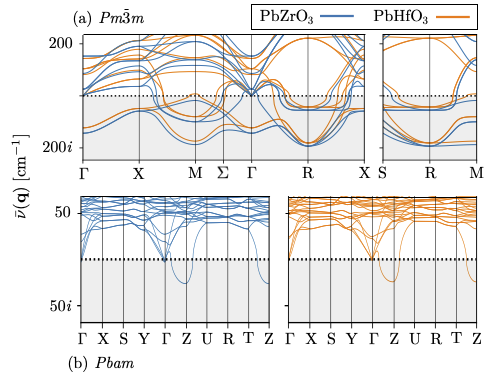
Unstable phonon modes in PbZrO3 and PbHfO3 (a) In the high temperature cubic paraelectric structures. (b) In the low temperature antiferroelectric structures. This sturcture is presently regarded as the ground state but clearly there is an instability near the Z-point in both materials.Most of my ventures into this area revolve around the study of phonon modes. In atomistic simulations, should phonon modes be found with imaginary frequencies, this indicates a dynamical instability in the crystal structure. Simply put, the the nuclear displacement patterns associated with the unstable modes define a potential symmetry lowering phase transiton pathway. After having noticed particularly long wavelength patterns with such a property in PbZrO3 (and the well-known piezoelectric PZT), we found structures of PbZrO3 and PbHfO3 more stable than their at-the-time accepted ground state phase.
Despite this, many of these structures are not observed in experiment which leaves us with the question "why?". One area I am investigating is the role of quantum effects in suppressing certain vibrational modes in antiferroelectrics. Another is the interplay of strain and strain gradients in stabilizing exotic displacement patterns. Many of these questions can be answered from a first principles DFT standpoint (with standard codes or otherwise) but some may require long duration molecular dynamics simulations with systems of millions of atoms. At this stage, it is most computationally efficient to look to second principles methods and first-principles derivied methods like machine learned interatomic potentials.